

The FDA Decision That Just Setback America's mRNA Vaccine Industry
The far reaching implication of Vinay Prasad's unprecedented refusal-to-file letter for Moderna's flu shot


If you haven’t checked the vibes in the room, the entire BioPharma community is in an uproar at Center for Biologics Evaluation and Research (CBER) director Vinay Prasad’s latest action. Per reports, Moderna MRNA 0.00%↑ received the rare refusal-to-file (RTF) letter from CBER after using a precious priority review voucher (PRV) for its mRNA-based influenza (flu) vaccine, mRNA-1010. It appears this wasn’t a group decision either. Prasad unilaterally decided to issue the RTF, while his reviewers were in support of accepting Moderna’s filing for review.
In the RTF (which Moderna released here), Prasad esentially says that Moderna’s application is incomplete because it lacks an appropriate comparator, namely that the main study used in Moderna’s filing package (called P304) examining adults ages 50 years old and older, was comparing Moderna’s drug to standard-dose flu vaccine, instead of the high-dose flu vaccine, which is recommended in use for the 65 and older group.
But Moderna is mad because they already had confirmation from the FDA that their design was acceptable, and further addressed questions about comparison to high-dose vaccines with supplementary Phase 3 immunogenicity data. Based on Moderna’s recounting of their interactions, they were given no reason to believe that their filing would not at least be reviewed. You usually don’t spend a PRV (which fetch $100-$200M on the open market) unless you felt high confidence in your application being accepted.
Prasad (and his boss Marty Makary, head of the FDA) defended the decision, effectively arguing that a high-dose comparator should have been used in the main study for 65+ age group because that is the current standard of care in the US (although not in much of the rest of the world).
This latest saga at the FDA involving Vinay Prasad seems like more of the same, but there are far reaching implications to this action that the BioPharma sector should consider.
A Major Setback for Vaccine Development and Investment
Moderna is left swimming up stream without a paddle. They ran multiple 40K+ size global studies all to have the FDA basically say their studies are effectively useless and not even worth considering. That’s obviously millions of dollars flushed down the drain and a steel vault door shut in the face of America’s leading mRNA manufacturer.
More importantly, it’s one less effective medicine available for patients, not just for this flu season (which has been quite severe) but many flu seasons to come. Moderna has come out and said that they aren’t even going to continue investing in P3 vaccine trials for infectious diseases in the US or anywhere else. While they will still likely take mRNA-1010 into international markets, it doesn’t make financial sense for the company to continue pursuing these sorts of vaccines if the US market is going to be off limits to them.

Of course some of this commentary is posturing by Moderna's CEO, but I don't blame him. Vaccines are unique in how heavily the US market subsidizes global access. American reimbursement rates and market size essentially bankroll the development and distribution pipelines that supply the rest of the world. Cut off US market access for mRNA vaccines, and the economics collapse for Moderna domestically and for every international market counting on these products. If Moderna follows through on retreating from infectious disease development, foreign regulators may not have as many mRNA vaccines to approve. The only salvage pathway would be out-licensing to partners willing to shoulder the clinical and commercial burden ex-US. This is a scenario that delays timelines, fragments supply chains, and ultimately could leave patients waiting longer for innovation.
Why should any other mRNA vaccine developers feel differently? Each of them, certainly smaller and less financial endowed than Moderna, is probably thinking the same thing. Is it worth continuing to pursue our infectious disease programs?
Investors too could be more likely to shy away from mRNA plays as well. The FDA’s shifting standard and lack of predictability adds immense late-stage to risk. No investor wants to fork over tens of millions of dollars only for their portfolio companies major inflection catalyst to be rendered moot because of a sudden unpredictable change in opinion at the FDA. This lack of consistency reverberates through all levels of the BioPharma ecosystem.
Miscommunications Abound
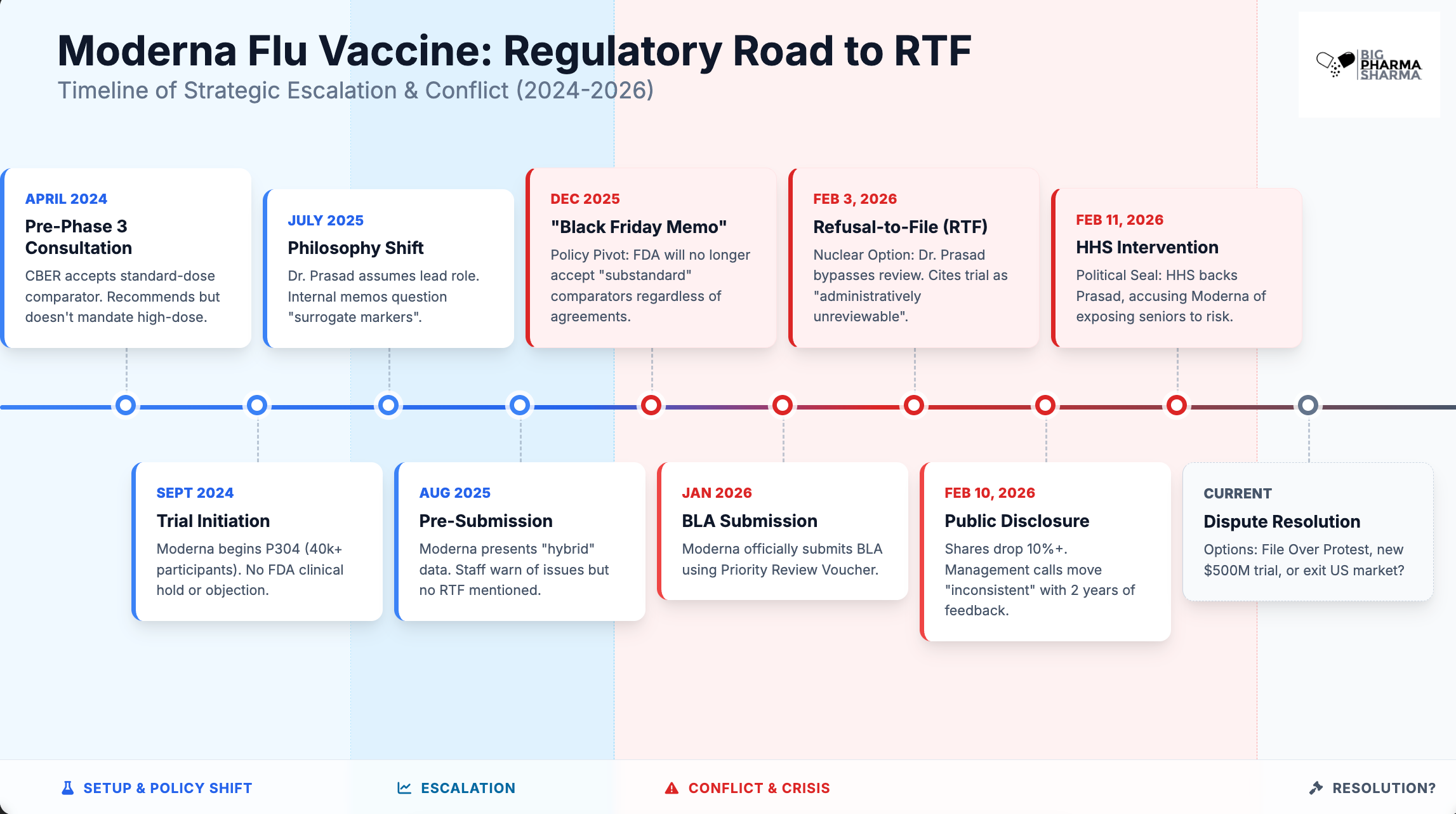
It is important to look at the timeline of events here to really understand what went wrong and why. And what stands out is a clear communication issue.
There is a disconnect between the higher ups at the FDA, like Prasad and Makary, and long-time staffers who work under them. From the outset in April 2024, the vaccine team at the FDA defines Moderna’s trial design as “acceptable” but “recommends” they use high-dose. Moderna opts to go the standard-dose route that was deemed “acceptable”. Prasad gets involved on this project after the study has already started (July 2025) and raises concerns. By August, FDA staff tell Moderna that the lack of a high-dose group in the control arm is going to be a “significant issue” (assuming that they got verbal guidance from Prasad on this here and are trying to flag that for Moderna) and requests additional analyses, which the company provided. This included data from a separate Phase 3 study comparing mRNA-1010 with a high-dose flu vaccine, but focusing on antibody levels rather than actual efficacy. Prasad has long been a critic of immunogenicity data as a substitute for outcomes in the approval of vaccines, and releases several internal FDA memos guiding the agency to move away from valuing these sorts of data points for approval.
